All published articles of this journal are available on ScienceDirect.

Impact of the N-terminal Fragment on the Solubility of eIF4E1 Solanum tuberosum
Authors Info & Affiliations
Abstract
Introduction
Obtaining soluble eukaryotic proteins using bacterial expression systems remains a significant challenge. Despite the availability of various techniques to optimize protein expression in E. coli, eukaryotic proteins are frequently expressed in an insoluble form when produced in prokaryotic cells.
Methods
Genes of interest were cloned into expression vectors, pET22b, and modified pET32a, using the restriction-ligation method. BL21(DE3), BL21(DE3) Star, BL21(DE3)pRARE, Tuner(DE3), Origami, SHuffle®T7, and C41(DE3) strains were used for analytical induction. Molecular modeling and molecular dynamics simulations were employed to design the experiments and to interpret the resulting data.
Results and Discussion
To prevent the accumulation of a truncated form of potato eIF4E in inclusion bodies, we tested various E. coli strains and repositioned affinity tags from the C-terminus to the N-terminus of the protein. Only the full-length eIF4E was found to be soluble in the prokaryotic expression system. Based on the eIF4E model and molecular dynamics simulations, we proposed a potential explanation for the impact of the N-terminal fragment on protein solubility.
Conclusion
The interaction between the N-terminal fragment and the dorsal surface of eIF4E may prevent protein aggregation. This shielding of hydrophobic regions appears to be a key factor in reducing aggregation, thereby facilitating the expression of eIF4E in a soluble form.
1. INTRODUCTION
The accumulation of proteins in inclusion bodies remains a significant challenge for many recombinant proteins produced in E.coli. Numerous protocols have been developed for the solubilization of proteins from inclusion bodies [1-5]. The isolation of proteins from inclusion bodies is a process of denaturation and their subsequent renaturation. Cotranslational protein folding is much more efficient than in vitro refolding. It is not clear whether the protein retains its functional activity after refolding or not. Additional tests should be performed to confirm the activity of the obtained protein. There are several reasons why proteins accumulate in inclusion bodies during synthesis, such as protein aggregation, improper formation of inter- or intramolecular disulfide bonds, toxicity of target proteins, and overexpression due to strong induction. Different strategies have been developed to obtain proteins in a soluble form [6-8].
The amino acid sequence at the N-terminus of a recombinant protein can determine its propensity for aggregation and accumulation in inclusion bodies. For example, charged amino acids may enhance protein solubility by forming hydrogen bonds, whereas hydrophobic residues tend to promote aggregation. To prevent the accumulation of protein in the inclusion bodies, various techniques can be used, for example, adding a signal peptide to the N-end of the protein [9, 10]. The signaling peptide promotes protein transport to a specific location in the cell. Various modifications at the N-terminal part, such as acetylation or methylation, can lead to changes in the interaction of the molecule with the solvent, which also affects the solubility of the protein [11-13]. However, since posttranslational modifications occur predominantly in eukaryotic systems, their relevance is limited in bacterial cells. The co-expression of target protein and N- or C-terminal tags, such as thioredoxin, SUMO, or MBP, can also affect the accumulation of proteins in soluble form [14-17]. Moreover, sometimes, the movement of the tag from the C to the N-terminus can have a positive effect on protein solubility [18-20]. The presence of 6His at the N- or C-terminus can affect the thermal stability of the protein, which is an important indicator when working with recombinant proteins [21].
During the heterologous expression of the translation initiation factor 4E (eIF4E) from Solanum tuberosum in a bacterial system, we encountered a major obstacle: the accumulation of the protein in inclusion bodies. eIF4E is a cap-binding protein that is involved in the initiation of translation in eukaryotic cells. It is а component of the eIF4F complex, which binds to the mRNA cap and attracts the 43S pre-initiation complex [22-25]. eIF4E is represented by a family of proteins. This family is one of the factors that makes Solanaceae plants susceptible to potyviruses. However, there are no experimentally determined structures of eIF4E proteins of Solanaceae. The interaction of proteins of the eIF4E from the Solanaceae family with VPg of potato Y virus is a necessary condition for the development of viral infection [26-28]. There is no experimental data confirming the mechanism of interaction between eIF4E and VPg, making structural studies crucial. The significant impact of potyviruses on crop yields also makes it an urgent priority to study the mechanism of potyviral RNA translation. Solving the structure of the eIF4E-VPg PVY complex would give an obvious interpretation of the previously obtained data, explain the mechanism of interaction between the two proteins, and predict mutations that could disrupt this interaction. We need to obtain a large amount of functionally active recombinant eIF4E for protein crystallization, as well as for functional studies. However, the tendency of eIF4E to accumulate in inclusion bodies makes obtaining active protein multistage and inefficient. To address this challenge, we have conducted analytical induction of different variants of potato protein eIF4E1 in various E. coli expression strains in order to obtain this protein in a soluble form.
2. MATERIALS AND METHODS
2.1. Nucleotide Sequences
The nucleotide sequence of cDNA eIF4E1 was used to create expression vectors (Genbank accession: MT828879).
2.2. Molecular Modeling and Molecular Dynamics
The eIF4E1-49aa model was obtained as previously described [29, 30]. Alphafold3 was used to obtain full-length eIF4E1 models [31].
Molecular dynamics studies of the obtained theoretical model of the full-size eIF4E1 were carried out using the Gromacs 2022.3 software package [31-34] and the all-atomic force field Charmm36 (July 2022 version: CHARMM all-atom force field (July 2022)) [35]. The solution was modeled using a three-point CHARMM TIP3P (TIPS3P) water model. The system was equilibrated to achieve a constant temperature of 310 K and a constant pressure of 1 bar. The molecular dynamic trajectory was calculated in a time interval of 200 ns at a given constant pressure and temperature with a time step of 2 fs. Manual editing of the models was carried out in the Coot program [36].
2.3. Data Analysis and Visualization
The superposition and comparative analysis of experimental and theoretical eIF4E models were carried out in the software packages Coot and PyMOL (Version 2.5.0a0. OPEN-SOURCE). Clustering and analysis of the molecular dynamic trajectory were performed in the Gromacs and VMD software packages [37]. Clustering of MD trajectory frames was carried out for the Cα atoms of the model using the gmx cluster module and the GROMOS algorithm, applying a threshold value of rmsd=1.5A. As a result, 81 clusters were obtained with a maximum occupancy of 6240 frames out of 20,000 frames of the trajectory. For a full-length eIF4E1 potato model, a molecular surface with coloring according to the Wimley-White ΔG water-membrane hydrophobicity scale [38] was constructed and visualized in the MolStar program [39]. Visualization of the obtained data was carried out using the Inkscape vector graphics program (https://inkscape.org/) and the software complexes of molecular graphics PyMOL and MolStar.
2.4. Plasmid Design
Sequences of eIF4E1 (1-696 b.p.) and eIF4E1-49aa (145-696 b.p.) were amplified from cDNA of the Solanum tuberosum cultivar Zhukovskiy ranniy. A modified pET32a-GG-ΔTrx vector was used for cloning. The thioredoxin located between the two NdeI sites was removed; the BsaI recognition site after the enterokinase site was also added to the plasmid, while preserving the triplet encoding alanine between the enterokinase site and the BsaI site. The amplicons and vector were treated with BsaIHF-v2 (NEB) restrictase and then ligated using a highly active T4 DNA ligase by the Golden Gate method (according to the manufacturer’s protocol). The introduction of restriction sites was carried out to obtain a more user-friendly vector. The nucleotide sequence of eIF4E1-49aa (145-696 b.p.) was subcloned into the expression vector pET22b at the restriction sites, NcoI and XhoI, before the sequence of the signal peptide. This signaling peptide is intended to facilitate the movement of the protein into the periplasmic space [9]. All plasmids were sequenced. Each of the resulting genetic constructions carries a 6His-tag to facilitate affine purification.
2.5. Analytical Induction and Protein Isolation
E. coli strains BL21(DE3) (Novagene), BL21(DE3) Star (Novagene), BL21(DE3)pRARE(Novagene) (Cm), Tuner(DE3) (Novagene), Origami (Novagene) (Kn+Tet), SHuffle®T7(Novagene) (Str), and C41(DE3) (Novagene) were transformed by plasmid pET22b-eIF4E1-49aa(Amp) and grown on the selective agar-media during 16 h +37 °C. One colony from the agar plate was transferred into 10 ml selective Luria-Bertani (LB) medium and incubated in a shaker at +37 °C 180 vol.\min. When the optical density of OD590 reached ≈0.6–0.8, the inductor was added to the final concentration of 0.5 mM isopropyl-β-D-thiogalactoside (IPTG). Cells were incubated during 3 h at +37 °C or 20 h at +20 °C. The expression level was checked using SDS-PAGE. The cells were precipitated by centrifugation and then resuspended in buffer (10 mM Tris HCL pH7.5. 250 mM NaCl, 10% glycerol) with the addition of DNase and PMSF. The cell suspension was homogenized by sonication. To avoid overheating during sonication, a container with a suspension of cells was placed into watered ice. After homogenization, the suspension was centrifuged and the presence of protein in soluble and insoluble fractions was determined by electrophoresis. The target protein was verified by mass spectrometry.
The calculation of the expected molecular weight of the protein, the coefficient of extinction, and the isoelectric point was carried out using the Biochemistry Online tool (https://vitalonic.narod.ru/biochem/algorithm.htm).
3. RESULTS AND DISCUSSION
3.1. Overview of Protocols of eIF4E Isolation for Crystallization
To begin with, we analyzed purification protocols of human, mouse, yeast, nematode, wheat, pea, and melon eIF4E, which were successfully crystallized [40-44]. All mentioned eIF4E proteins were N-terminally truncated: 1-51aa (amino acids) were removed in melon eIF4E (UniProtKB Q00LS8), 1-50 aa were removed in pea eIF4E (UniProtKB Q0GRC4), 1-36 aa were removed in wheat eIF4E (UniProtKB P29557), 1-28 aa were removed in mouse eIF4E (UniProtKB P63073), and 1-30 aa were removed in nematode eIF4E (UniProtKB O61955). Purification protocols for these proteins are very similar. The E. coli strain Rosetta (DE3) pLysS (Novagen) was used for the expression of melon, pea, and wheat eIF4E, and the strains HB101(DE3) and BL21(DE3) Star were used for the expression of mouse and nematode eIF4E, accordingly. Expression strains with plasmids that encode rare tRNAs are often used to induce the synthesis of eukaryotic proteins in the prokaryotic system. All protocols used LB medium, IPTG at a concentration between 0.4 and 0.6 mM, and a sonication or a French press as the homogenization method. Nematode and melon eIF4E were coexpressed with the eIF4G fragment. Accumulation of the target protein in inclusion bodies and its isolation under denaturing conditions was reported only for the mouse N-terminally truncated eIF4E. Renaturation of this protein does not prevent its crystallization [42, 45].
3.2. eIF4E Modeling and Design of Expression Vectors
Mobile elements in the structure of proteins often prevent their crystallization. The N-terminal fragment, which was deleted in the mentioned eIF4E, is unstructured and mobile. It was shown in the structure of full-length eIF4E obtained in the NMR experiment (PDB code: 4B6U). The absence of this fragment in the recombinant protein should not affect its functional activity or the integrity of the globular part, but at the same time, the possible role of this fragment in the interaction of eIF4E with eIF4G or other molecules cannot be excluded.
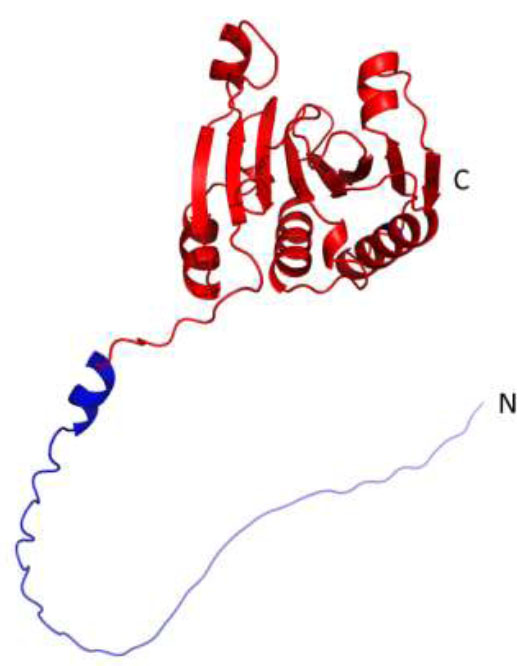
The model of Solanum tuberosum eIF4E1. The blue color indicates the N-terminal fragment (1-48 aa), which is missing in the shortened version of the protein (eIF4E1-49aa).
In this regard, we also started working with a shortened version of eIF4E1 for the following crystallization. Using the previously obtained model, we determined the boundaries of the N-terminal mobile unstructured part and the globular part of eIF4E1: 1-48 and 49-233, accordingly (Fig. 1). The shortened version of eIF4E is referred to as eIF4E1-49aa in the following text. At the same time, for functional experiments, we needed both a full-length and a shortened version of the target protein. Therefore, we obtained several variants of vectors encoding both the full-length and shortened versions of eIF4E.
The sequences encoding eIF4E1-49aa and full-length eIF4E1 were cloned into the expression vector pET32a-GG-ΔTrx. As a result, pET32a-GG-ΔTrx-eIF4E1-49aa and pET32a-GG-ΔTrx-eIF4E1 plasmids were obtained. Using the first plasmid enabled us to get the target protein with an estimated molecular weight of 25.8 kDa. This variant includes a 6His-tag, thrombin cleavage site, S-tag, and enterokinase cleavage site at the N-terminus (Fig. 2). Additionally, using the pET32a-GG-ΔTrx-eIF4E1 plasmid allowed us to get the target protein with an estimated molecular weight of 30.8 kDa. This variant of the protein contains the same N-terminal sequence as previously mentioned (Fig. 2). This design allows the removal of the tag during one of the purification steps.
For the expression of full-length eIF4E1 and eIF4E1-49aa, we used the BL21(DE3)-pRARE strain that has an additional plasmid encoding rare tRNAs [46]. One potential cause of protein accumulation in inclusion bodies is the misincorporation of amino acids, which can result in improper folding. Expression of full-length eIF4E1 in this strain during 3 h at +37 °C and 20 h at +20 °C led to the formation of a soluble form of full-length eIF4E1 (Fig. 3). In contrast, under the same conditions, eIF4E1-49aa accumulated in inclusion bodies.
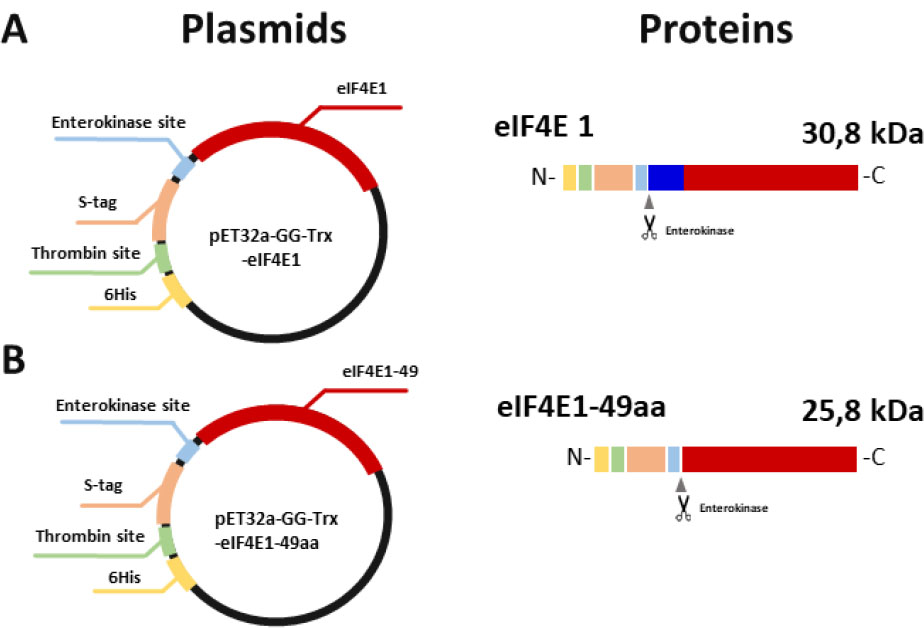
Schematic representation of the expression plasmids and the primary sequence of the target proteins. Different colors on the circle indicate the main elements, located between the T7 promoter and the terminator. The same colors indicate the corresponding elements on the primary structure of the protein. Scissors indicate the place where the protease cut the protein. A – Scheme of the plasmid encoding full-length eIF4E1 and the protein primary sequence; B – Scheme of the plasmid encoding eIF4E1-49aa and the protein primary sequence.
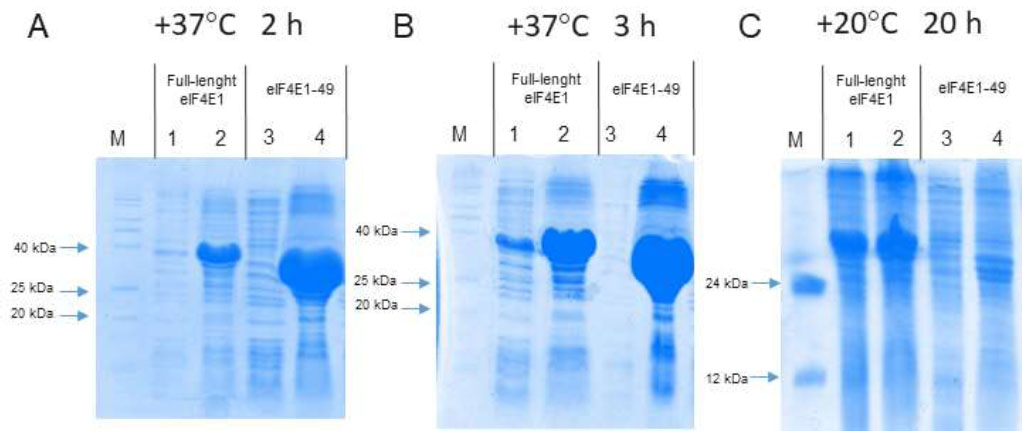
Electrophoregrams of the samples obtained after analytical induction in the strain BL21(DE3) pRARE. Odd numbers correspond to samples of supernatant after cell sonication; even numbers correspond to samples of debris after cell sonication. 1 and 2 correspond to full-length eIF4E1; 3 and 4 correspond to eIF4E1-49aa; M – protein molecular weight markers. A – protein expression during 2 h at +37 °C; B – protein expression during 3 h at +37 °C; C – protein expression during 20 h at +20 °C.
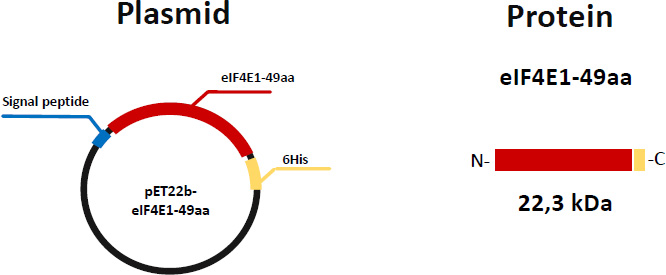
Schematic representation of the expression plasmid pET22b and the primary sequence of eIF4E1-49aa. Different colors on the circle indicate the main elements, located between the T7 promoter and the terminator. The same colors indicate the corresponding elements on the primary structure of the protein.
Isolation of proteins from inclusion bodies is not an impossible task. However, this complicates the isolation process and does not guarantee that the target protein will refold correctly and will be functionally active. In this regard, we next modified the genetic construct to obtain eIF4E1-49aa in a soluble form. Since the presence of additional tags (despite the possibility of getting rid of them) is undesirable for crystallization experiments, we selected the expression vector pET22b, which does not code sequences of long tags and sites for protease digestion. The sequence encoding eIF4E1-49aa was cloned into this vector. As a result, the pET22b-eIF4E1-49aa plasmid was obtained. This construction enabled us to get the target protein with an estimated molecular weight of 22.3 kDa. This variant includes a 6His-tag at the C-terminus and 22 aa signal peptide at the N-terminus, which is expected to help the protein to be released into the periplasmic space (Fig. 4).
In our attempts to get soluble SteIF4E1-49aa, we tried different E. coli strains and growth conditions. Analytical induction of synthesis of eIF4E1-49aa from pET22b plasmid in strains BL21(DE3) and BL21(DE3)-pRARE during 3 h at +37 °C and 20 h at +20 °C did not lead to the production of the soluble protein (Fig. 5).
Lowering the temperature slows down the process of protein synthesis, reducing the likelihood of translational errors and potentially improving protein solubility. However, in our case, it was ineffective. Another factor contributing to strong protein aggregation and, consequently, its accumulation in inclusion bodies is the presence of unpaired cysteine residues in the protein structure. The Shuffle and Origami E. coli strains were created to improve the accuracy of the formation of disulfide bonds and were also selected for our experiment [47, 48]. We also used the E. coli strain C41(DE3), which is recommended for the expression of toxic proteins, and the strain Tuner (DE3), which is suitable for toxic and insoluble proteins. Unfortunately, all these strains did not yield a positive result for the short version of SteIF4E1 (Fig. 5).
The full-length version of eIF4E1 was produced in soluble form under standard conditions (Table 1) without the need for further optimization. The presence of any tags at the N- or C-end of the eIF4E1-49aa, lowering of the incubation temperatures, and using different E. coli strains had no positive effect on the solubility of eIF4E1-49aa (Table 1).
The sequence of SteIF4E1 N-terminal part (1-48 aa) (MATAEMERTTSFDAAEKLKAADAGGGEVDDELEEGEIVEESNDTASYL) does not show any similarity with any known signaling peptides sequence. At the same time, experimental data suggest that the N-terminal fragment (1-48 amino acids) protects the protein from aggregation.
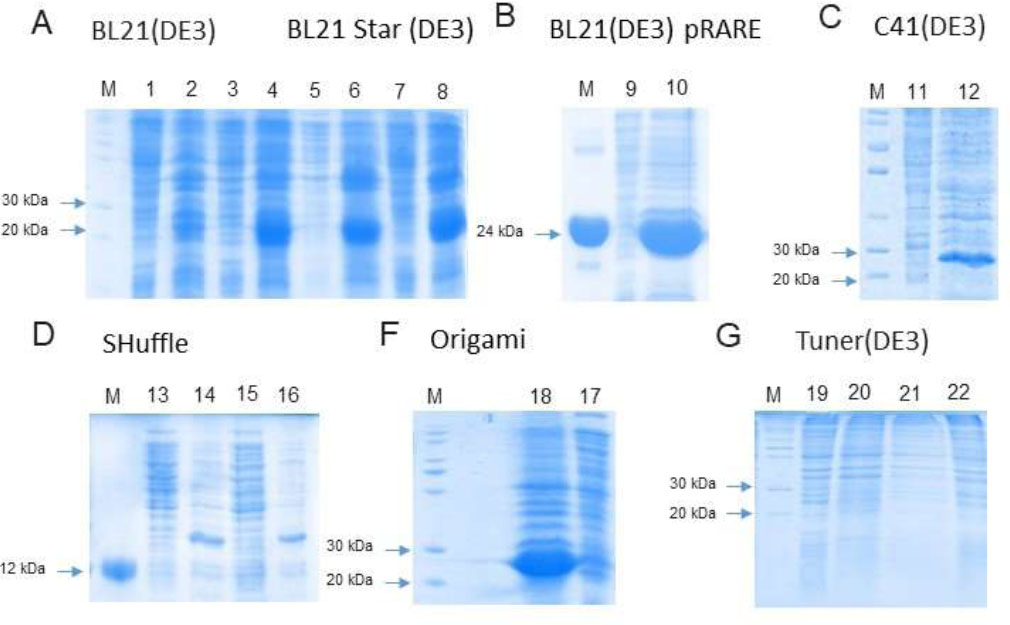
Electrophoregrams of the samples obtained after analytical induction of the eIF4E1-49aa in different strains. Odd numbers correspond to samples of supernatant after cell sonication; even numbers correspond to samples of debris after cell sonication. M – protein molecular weight markers. A – 1, 2, 3, 4 – strain BL21(DE3): 1, 2 – protein expression during 3 h at +37 °C, 3, 4 – protein expression during 20 h at +20 °C. 5, 6, 7, 8, – strain BL21(DE3)Star: 5, 6 – protein expression during 3 h at +37 °C, 7, 8 – protein expression during 20 h at +20 °C, B – strain BL21(DE3) pRARE: 9, 10 – protein expression during 20 h at +20 °C; C – strain C41(DE3), 11, 12 – protein expression during 20 h at +20 °C. D – strain Shuffle: 13, 14 – protein expression during 3 h at +37 °C, 15, 16 – protein expression during 20 h at +20 °C. E – strain Origami: 18, 17 – protein expression during 20 h at +20 °C. F – strain Tuner(DE3): 19, 20 – protein expression during 3 h at +37 °C, 20, 21 – protein expression during 20 h at +20 °C.
| Protein | Vector | Strain | Result | ||
|---|---|---|---|---|---|
| 37 °C 3 h | 20 °C 20 h | ||||
| eIF4E1-49aa | pET22b | BL21(DE3) | Insoluble | Insoluble | |
| BL21(DE3) pRARE | Insoluble | Insoluble | |||
| BL21(DE3)Star | Insoluble | Insoluble | |||
| C41(DE3) | - | Insoluble | |||
| SHuffle | There is no induction | ||||
| Origami | - | Insoluble | |||
| Tuner (DE3) | There is no induction | ||||
| eIF4E1-full | pET32a-GG-ΔTrx | BL21(DE3) pRARE | Soluble | Soluble | |
| eIF4E1-49aa | pET32a-GG-ΔTrx | BL21(DE3) pRARE | Insoluble | Insoluble | |
3.3. Structural Interpretation of the Influence of the N-Terminal Fragment on Protein Solubility
These data can be interpreted from a structural point of view using molecular modeling and molecular dynamics methods. A comparative analysis of potato eIF4E1 and homology protein models, both experimental and theoretical (including those generated using the latest version of the Alphafold3 algorithms), revealed the ability of different regions of the unfolded N-terminal fragment to form alpha helices.
According to the results of molecular modeling and molecular dynamics studies, this fragment is able to interact with the globular part of the protein, forming stable contact with a section of its dorsal surface (Fig. 6). The dorsal surface of eIF4E forms part of the eIF4G binding site and is rather hydrophobic. The exposition of such kind of hydrophobic surfaces to a solvent (which is the case for the models with the truncated N-terminal part) could cause the aggregation of proteins in a water solution. The cluster analysis of the 200ns MD trajectory for the intact factor gives us the most stable conformation for the N-terminal part in this experiment. This example shows the ability of the N-terminal fragment to shield the hydrophobic region of the eIF4E globule surface, extending the area of the hydrophilic one. This could be the reason for the increased solubility of the intact protein compared to the truncated one. Thus, the above-mentioned interaction of a part of the N-terminal fragment with the eIF4E dorsal surface can prevent aggregation, preserving the protein in a soluble form.
If we take a look at the same region of eIF4E from other organisms, we will also find the hydrophobic spot (Fig. 1S). The intensity of these regions can explain the tendency of the proteins to aggregate.
A. Cartoon representation of the most stable conformation of the model of full-length eIF4E1 from the 200ns molecular dynamic trajectory. The view from the cap-recognizing loop is on the left. The view from the eIF4G binding site is on the right. The gray color shows the globular part of the factor. The green color shows the N–terminal fragment of the factor, which was initially poorly structured and adopted a conformation during the MD experiment.
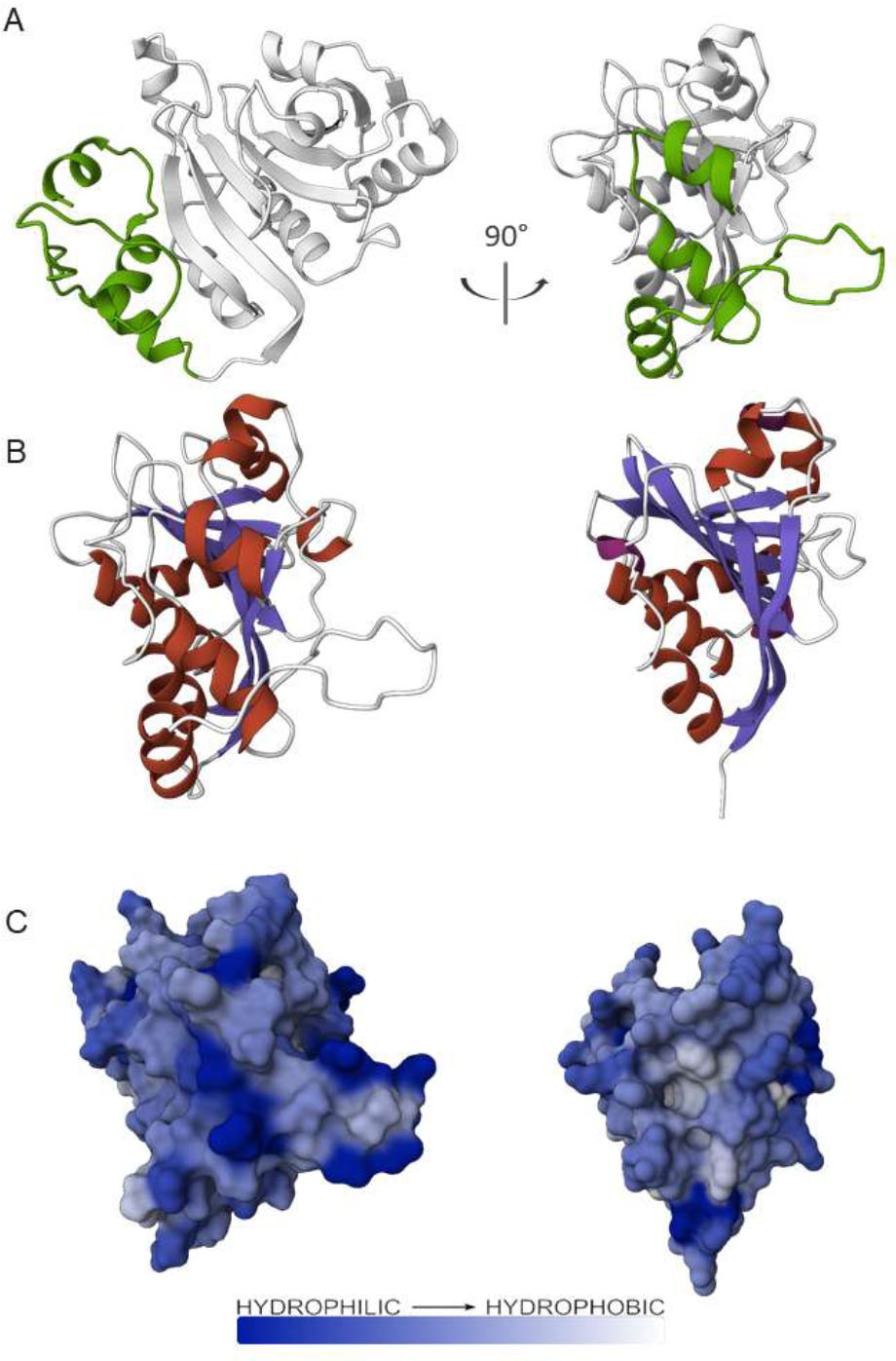
Possible role of the N-terminal fragment in the extension of the hydrophilic surface of eIF4E1.
B. Cartoon representation of the models of full-length eIF4E1 (left) and eIF4E1-49aa (right), viewed from the back side. The elements of the secondary structure are colored: α-helixes are highlighted in red, β-strands are highlighted in slate, and loops are highlighted in white.
C. Surface representation of the models of full-length eIF4E1 (left) and eIF4E1-49aa (right) from the dorsal view. The surfaces are colored according to the Wimley-White ΔG water-membrane hydrophobicity scale [38].
CONCLUSION
The full-size version of eIF4E1 obtained in bacterial cells is soluble. The shortened version of eIF4E1 (eIF4E1-49aa) accumulates in inclusion bodies. The presence of a hydrophobic region on the surface of the molecule provides a tendency for the protein to aggregate. This is due to the fact that the N–terminal fragment of the full-size eIF4E1 variant interacts with the hydrophobic surface of the globule, which makes it more hydrophilic, thereby preventing aggregation. Using the method of molecular modeling and molecular dynamics, it is possible to identify such hydrophobic interface regions and find a way to shield them. This approach will make it possible to obtain aggregation-prone proteins containing hydrophobic regions in a soluble form.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: E.Y.N.: Study conception and design; V.V.K. and V.Y.K.: Data collection; P.T.D.: Methodology; O.S.N.: Analysis and interpretation of results; and V.V.A.: Visualization. All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| LB | = Luria-Bertani |
| IPTG | = Isopropyl-β-D-thiogalactoside |
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information are available within the article.
FUNDING
The work was carried out with the financial support of the Russian Science Foundation: grant No. 24-44-04007.
ACKNOWLEDGEMENTS
The authors would like to thank the Plant Stress Tolerance Laboratory of the All-Russia Research Institute of Agricultural Biotechnology for collaboration.
SUPPLEMENTARY MATERIAL
Fig. (1S). Surface representation of a few members of the eIF4E family from the dorsal view.
The surfaces are colored according to the Wimley–White ΔG water-membrane hydrophobicity scale [38]. A – Crystal structure of eIF4E from Cucumis melo; B – Crystal structure of eIF4E from Drosophila melanogaster; C – Crystal structure of eIF4E from Mus musculus; D – Crystal structure of eIF4E from Pisum sativum.

